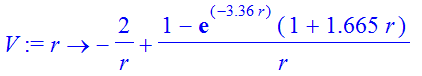
Atomic Model
A simple model for the structure of few-electron atoms is presented. A model potential for the helium atom is introduced (it represents a good approximation to the so-called Hartee-Fock potential for the helium atom). The eigenenergies and eigenfunctions of this potential are determined by
a) the variational method for the lowest-lying eigenstate for a given angular momentum symmetry;
b) the numerical solution of the radial Schroedinger equation;
c) a matrix diagonalization using a Slater-type orbital basis.
A simple model potential for the ground state of helium can be obtained as follows: assume that in the ground state the wavefunction corresponds to an 1s^2 configuration (one spin-up, one spin-down electron to form parahelium with total electron spin zero). What potential should the electron experience?
The nuclear attraction -2/r and a repulsive potential due to the presence of the other electron.
What is the repulsive potential due to the other electron (which has the same 1s-wavefunction, it has just opposite spin projection)?
We have an chicken-and-egg problem here: given a potential, we can determine the |1s> state, and calculate the repulsive part of the potential correctly. This is the objective of a self-consistent field calculation. Then we would need to repeat the calculation of the new |1s> state using the new potential. Repeating this procedure one can come up with a |1s> state and corresponding potential that provide the lowest energy.
We break the closed circuit by stating an approximate answer for the potential in helium:
| > | restart: Digits:=14: |
| > | V:=r->-2/r+1/r*(1-exp(-3.36*r)*(1+1.665*r)); |
This is the potential experienced by an 1s-electron in the helium atom: at short distances the electron just feels the full nuclear attraction (-2/r) as the second expression approaches a constant as r goes to zero. At large distances the electronic repulsion term goes like 1/ r and screens the nucleus by one unit so that the overall potential goes like -1/ r .
We can check for the consistency by a simple variational calculation:
We start with an unnormalized 1s state that depends on a 'charge' parameter (cf. the hydrogen-like wavefunction)
| > | chi:=r*exp(-beta*r); |
![]()
We are using the radial wavefunction (i.e. r*R_nl(r) ) so that the radial kinetic energy is just proportional to the second derivative.
| > | assume(beta>0); |
| > | E1sT:=Int(chi*(-1/2*Diff(chi,r$2)+V(r)*chi),r=0..infinity)/Int(chi^2,r=0..infinity); |
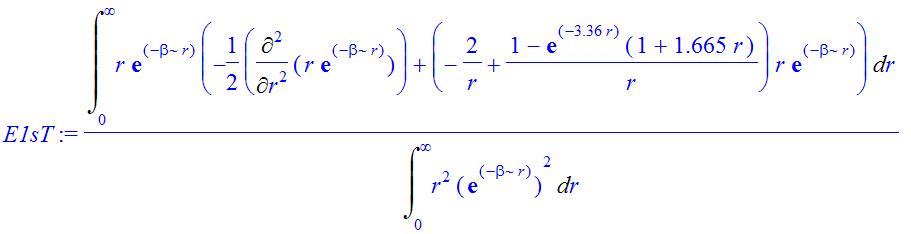
| > | E1sT:=value(E1sT); |
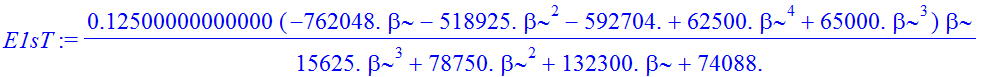
| > | plot(E1sT,beta=1.55..1.8,thickness=3); |
![[Maple Plot]](images/AtomicModel5.gif)
| > | beta0:=fsolve(diff(E1sT,beta),beta=1..2); |
![]()
| > | E0:=subs(beta=beta0,E1sT); |
![]()
Note that the beta-value is less than 2. The value of beta=2 would be obtained if there was only one 1s-electron (the hydrogen-like solution for the ground state of the He+ ion). The fact that the electron wants to have a slightly more diffuse wavefunction reflects the so-called inner screening: the electrostatic repulsion when combined with the simple model of two identical electrons (apart from the spin projection) leads to two electrons which repel each other on average (independent electron model). Both electrons experience the same common central potential and are bound by the same eigenenergy.
Now we would like to check two things:
1) how accurate is this variational solution?
2) how close is the used potential to the potential produced by this 1s-wavefunction?
Part 1 can be answered by using dsolve[numeric]. For part 2 we need to solve the Poisson equation.
Let us start with the first question: To solve the SE we start the integration not at zero, but at some small value of r=eta to avoid the singularity.
| > | eta:=10^(-8); |
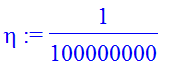
| > | IC:=phi(eta)=eta,D(phi)(eta)=1; |
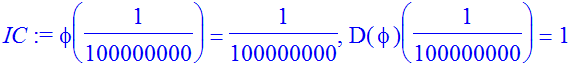
| > | Et:=-0.9042; |
![]()
| > | SE:=-1/2*diff(phi(r),r$2)+(V(r)-Et)*phi(r); |
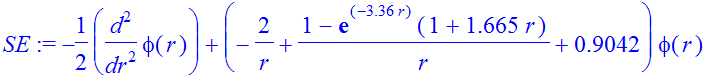
| > | sol:=dsolve({SE,IC},phi(r),numeric,output=listprocedure): |
| > | phir:=subs(sol,phi(r)): |
| > | plot(phir,0..10,thickness=3); |
![[Maple Plot]](images/AtomicModel12.gif)
| > | E1s:=Et; |
![]()
The numerically exact solution for the radial SE yields a slightly lower eigenvalue of E_1s = -0.9042 a.u. (1 a.u. = 27.21 eV) compared to the variational result of E_1s_v =-0.897 a.u.. This means that the true eigenfunction is somewhat different from the hydrogenic form.
The numerical eigenfunction is not normalized properly. To compare the graphs we simply change the normalization of the variational answer. In fact, the variational state was normalized such that the derivative of the function equals unity at r =0.
| > | eval(subs(r=0,diff(subs(beta=beta0,chi),r))); |
![]()
| > | with(plots): |
Warning, the name changecoords has been redefined
| > | P1:=plot(phir,0..6,color=red,thickness=3): |
| > | P2:=plot(subs(beta=beta0,chi),r=0..6,color=blue,thickness=3): |
| > | display([P1,P2]); |
![[Maple Plot]](images/AtomicModel15.gif)
Note that the agreement for the energy was at the level of 7/900 , i.e. in the 1 % range. The deviation between the wavefunctions is in the 10 % range.
Nevertheless, we can state that the simple hydrogenic wavefunction catches the main feature of the numerical solution, namely the most likely location in r for the 1s electron.
The result for the binding energy for one of the two helium electrons is the first quantity that can be compared with experiment:
The ionization potential of helium is measured to be 24.481 eV (cf. R. Liboff, table 12.2). Our answer is:
| > | -Et*27.21*_eV; |
![]()
For any atom the eigenenergy of the highest occupied orbital should equal the negative of the ionization potential. Our result is indeed quite close.
There is another quantity that can be measured, namely the total energy of the atom (equal to the sum of ionization energies for both electrons). This is not simply twice the eigenenergy, since after ionizing on of the two He-electrons, the other is left in a hydrogen-like state (energy of -2 a.u. due to Z=2, and E_1s=-Z^2/2).
Combining this answer with the calculated 1s binding energy we have for the total binding energy:
| > | (Et-2)*27.21*_eV; |
![]()
The electrostatic repulsion:
Now we look at the question as to what potential is associated with the approximate wavefunction. For this we need a solution to the Poisson equation for a spherically symmetric charge distribution based on the properly normalized variational wavefunction.
| > | A1s:=1/sqrt(int(chi^2,r=0..infinity)); |
![]()
| > | rho:=subs(beta=beta,(A1s*chi)^2); |
![]()
| > | int(rho,r=0..infinity); |
![]()
Note that the 4Pi from the integration over theta and phi are cancelled by the square of the spherical harmonic Y00!
The solution to Poisson's equation in multipole expansion leads to the monopole expression:
| > | V0:=unapply(simplify(int(rho,r=0..R)/R+int(rho/r,r=R..infinity)),R); |
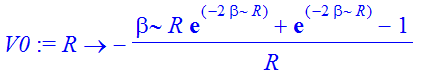
| > | plot([V(r)+2/r,subs(beta=beta0,V0(r))],r=0..5,color=[red,blue],view=[0..5,0..2],thickness=3); |
![[Maple Plot]](images/AtomicModel22.gif)
We recognize that the electronic repulsion in our helium atom of independent electrons is modelled after the simple hydrogen-like solution to the problem. The above potential shows how asymptotically the charge distribution of electron 1 screens one of the protons for electron 2 (located at a large r -value).
A sophisticated central-field or Hartree-Fock calculation take the electrostatic repulsion due to the numerically obtained charge density and re-calculates the eigenenergy/eigenfunction until convergence is achieved.
Electronic excitations
We proceed to calculate the energy levels for the 2s and 2p states. We simply assume that we can use the potential obtained for the ground state, and calculate the energy spectrum for this potential. For the 2s-state we will not carry out a variational calculation as the energy will not be guranteed to be above the exact eigenenergy for the given potential. We repeat our trial-and-error procedure to find a radial function with one node at r >0:
| > | IC:=phi(eta)=eta,D(phi)(eta)=1; |
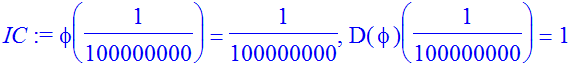
| > | Et:=-0.15768; |
![]()
| > | SE:=-1/2*diff(phi(r),r$2)+(V(r)-Et)*phi(r); |
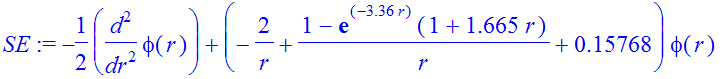
| > | sol:=dsolve({SE,IC},phi(r),numeric,output=listprocedure): |
| > | phir:=subs(sol,phi(r)): |
| > | plot(phir,0..20,-0.5..0.5,thickness=3); |
![[Maple Plot]](images/AtomicModel26.gif)
| > | E2s:=Et; |
![]()
How well does this agree with experiment? The National Institute of Standards (NIST) website in the US lists spectroscopic data using wavenumbers in cm^(-1). Relative to the ground state (1s^2) it lists for parahelium:
| > | dE1s2s:=166277.4*icm; |
![]()
| > | dE1s2p:=171134.9*icm; |
![]()
| > | dE1s3s:=184864.8*icm; |
![]()
The conversion factor is:
| > | eV:=8065.541*icm; |
![]()
| > | dE1s2s/eV; |
![]()
| > | dE1s2p/eV; |
![]()
Our 2s excitation energy in eV:
| > | (E2s-E1s)*27.21; |
![]()
The experimental difference between parahelium 1s2s and 1s2p states:
| > | (dE1s2p-dE1s2s)/eV; |
![]()
| > |
Now let us carry out the calculation for the 2p state. First we carry out the hydrogen-like variational calculation.
| > | l:=1; |
![]()
The trial function is hydrogen-like: the radial function picks up a factor of r^l, and we add the centrifugal potential to the radial Hamiltonian:
| > | chi:=r^(1+l)*exp(-beta*r); |
![]()
| > | E2pT:=Int(chi*(-1/2*Diff(chi,r$2)+(V(r)+1/2*l*(l+1)/r^2)*chi),r=0..infinity)/Int(chi^2,r=0..infinity); |
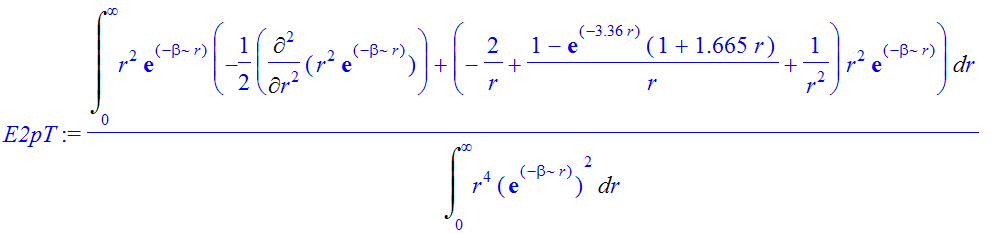
| > | E2pT:=value(E2pT); |
![]()
![]()
![]()
| > | plot(E2pT,beta=0.45..0.6,thickness=3); |
![[Maple Plot]](images/AtomicModel42.gif)
| > | beta0:=fsolve(diff(E2pT,beta),beta=0..2); |
| > | E2pV:=subs(beta=beta0,E2pT); |
![]()
![]()
Now we verify the variational calculation by a numerical solution:
| > | IC:=phi(eta)=eta^(l+1),D(phi)(eta)=(l+1)*eta^l; |
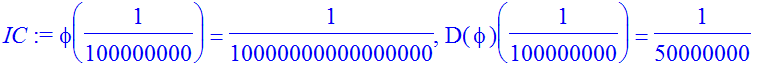
| > | Et:=-0.12699; |
![]()
| > | SE:=-1/2*diff(phi(r),r$2)+(V(r)+1/2*l*(l+1)/r^2-Et)*phi(r); |
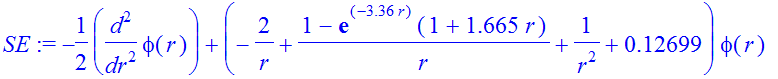
| > | sol:=dsolve({SE,IC},phi(r),numeric,output=listprocedure): |
| > | phir:=subs(sol,phi(r)): |
| > | plot(phir,0..25,thickness=3); |
![[Maple Plot]](images/AtomicModel48.gif)
| > | E2p:=Et; |
![]()
The answer is just slightly below the value obtained by the variational calculation with a hydrogenic wavefunction.
| > | (E2p-E1s)*27.21*_eV; |
![]()
We learn that the model predicts an excitation energy of slightly more than 21 eV. What does this have to do with reality?
This compares well with the experimental observation of parahelium (about 21.2 eV).
One important piece of information that does emerge from this calculation is the lifting of the ( nl )-degeneracy observed in the hydrogen atom spectrum: the 2s and 2p states are no longer degenerate, and thus, there is the possibility of observing photons that correspond to 2p-2s de-excitations.
How well do we get the splitting?
| > | (E2p-E2s)*27.21; |
![]()
This should be compared with the experimental result of 0.6 eV!
Matrix representation
We proceed with the calculation of approximate spectra in an angular momentum symmetry sector. For this purpose we first define ourselves a suitable basis. We defined so-called Slater-type orbitals for a given l -symmetry, and use a Gram-Schmidt procedure to orthogonalize them.
Our radial functions are real-valued, and therefore we have the simple inner product and normalization:
| > | IP:=(f,g)->int(f*g,r=0..infinity): |
| > | NO:=xi->xi/sqrt(IP(xi,xi)); |

| > | STO:=(n,l,beta)->r^(n+l)*exp(-beta*r); |
![]()
| > | L:=0; |
![]()
| > | N:=8; |
![]()
| > | B1:=[seq(STO(n,L,17/10),n=1..N)]; |
![B1 := [r*exp(-17/10*r), r^2*exp(-17/10*r), r^3*exp(-17/10*r), r^4*exp(-17/10*r), r^5*exp(-17/10*r), r^6*exp(-17/10*r), r^7*exp(-17/10*r), r^8*exp(-17/10*r)]](images/AtomicModel56.gif)
These states are linearly independent, but not orthogonal!
| > | IP(B1[1],B1[2]); |
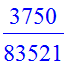
The Gram-Schmidt procedure takes a list of functions (state vectors) and orthonormalizes them using the two procedures.
| > | GS:=proc(vecs) local i,n,j,res,xi; |
| > | n:=nops(vecs); res:=[NO(vecs[1])]; |
| > | for i from 2 to n do: |
| > | xi:=vecs[i]-add(IP(vecs[i],res[j])*res[j],j=1..i-1); |
| > | res:=[op(res),NO(xi)]; od: end: |
| > | B1ON:=GS(B1): |
| > | IP(B1ON[5],B1ON[5]); |
![]()
| > | IP(B1ON[4],B1ON[5]); |
![]()
| > | B1ON[2]; |
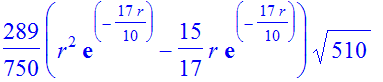
| > | plot([seq(B1ON[i],i=1..4)],r=0..10,color=[red,blue,green,black],thickness=3); |
![[Maple Plot]](images/AtomicModel61.gif)
Now we recycle the code from MatrixRep.mws to generate the Hamiltonian matrix: we define procedures to calculate the kinetic and potential energy matrix elements:
| > | Tkin:=(phi,psi)->-1/2*int(expand(phi*diff(psi,r$2)),r=0..infinity); |
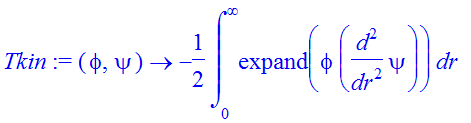
| > | Vpot:=(phi,psi)->int(expand(phi*psi*(V(r)+L*(L+1)/(2*r^2))),r=0..infinity); |
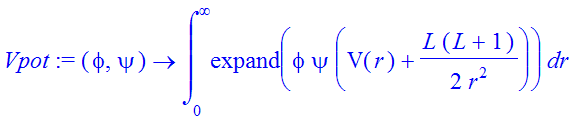
| > | with(LinearAlgebra): Digits:=15: |
| > | HM:=Matrix(N,shape=symmetric): |
We make use of the symmetry of the hamiltonian matrix: it allows to save almost a factor of 2 in computation time.
| > | for i from 1 to N do: for j from 1 to i do: HM[i,j]:=Tkin(B1ON[i],B1ON[j])+Vpot(B1ON[i],B1ON[j]); od: od: |
| > | #for i from 1 to N do: for j from i+1 to N do: HM[i,j]:=HM[j,i]: od: od: |
| > | HMf:=Matrix(evalf(HM),shape=symmetric,datatype=float[8]): |
| > | evals:=Eigenvalues(HMf, output='list'): |
| > | ev_s:=evalf(evals,8); |
![]()
| > | (-.90419974+.15516126)*27.21; |
![]()
We see that the chosen basis set can reproduce the 1s and 2s states, but that it does not have a prediction for a bound 3s eigenstate.
The 1s-2s energy difference can be compared with the experimental result for parahelium. We calculate 20.38 eV as compared to 20.61 eV in experiment. From the numerical solution we had 20.31 eV. Is the matrix size sufficiently large?
Exercise 1:
Increase the matrix size and observe the stability of the 1s and 2s eigenvalues. Do you find an acceptable 3s eigenvalue?
Exercise 2:
Change the value of the parameter that controls the Slater type orbital (STO) basis from the chosen value of 17/10. Find the best basis for the 3s eigenvalue.
Now the eigenfunctions. We use a symmetric matrix diagonalization now, and so the eigenvalues come out as reals and properly sorted. The eigenvectors are stored as a matrix, as a second entry in the sequence of expressions returned by Eigenvectors:
| > | VEs:=Eigenvectors(HMf): |
| > | VEs[1],VEs[2]; |

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Suppose that we would like to see the eigenfunctions corresponding to the four lowest-lying eigenvalues. The eigenvector for a given eigenvalue contains the expansion coefficients for the expansion of the eigenstate in terms of the chosen basis states.
The expansion coefficients are stored in columns. We pick the column index for a given state i , and then step through the row index j to assemble the states.
We have incorporated an explicit normalization of the assembled eigenfunctions even though this isn't necessary, as shown by the calculated value of No (it turns out to be practically equal to one). For an orthonormal basis (created by the Gram-Schmidt), and matrix eigenvectors normalized according to the Euclidean norm (usually adhered to by numerical diagonalization routines) this is true that normalized eigenfunctions are created automatically by the procedure.
| > | for i from 1 to 4 do: |
| > | psi0:=add(VEs[2][j,i]*B1ON[j],j=1..N): |
| > | No:=1/sqrt(int(expand(psi0^2),r=0..infinity)); |
| > | phi_a[i]:=No*add(VEs[2][j,i]*B1ON[j],j=1..N): od: |
| > | plot([seq(phi_a[i],i=1..4)],r=0..15,color=[red,blue,green,black],thickness=3); |
![[Maple Plot]](images/AtomicModel83.gif)
| > | No; |
![]()
The basis functions do not have a good span at large r where the higher states wish to reside. A smaller value of beta should improve matters.
For this reason one mixes usually STO's with different values of beta. The Gram-Schmidt process takes care of the orthonormalization.
Exercise 3:
Calculate the eigenfunctions for a few low-lying eigenstates in the L=2 and L=3 symmetry sectors. Observe how the eigenenergies approach the hydrogenic result in this case of E_n=-1/(2n^2). Can you explain this behaviour? Hint: graph the effective potential, and compare with that of a hydrogen atom.
| > |